Back to Whitepapers
Medical
AI in SDR
Scaling Security Design Reviews in Medical Device Companies - A Modern, Compliant Approach
Automate Security Design Reviews for medical devices. Meet FDA cybersecurity expectations with audit-ready, DHF-traceable threat modeling at scale.
By: Team Seezo
Executive Summary
Medical devices are now software-defined systems. Cybersecurity failures directly impact patient safety, product reliability, and regulatory outcomes. Connectivity, cloud services, mobile applications, and third-party dependencies have expanded the attack surface far beyond the physical device.
As a result, the U.S. Food and Drug Administration (FDA) expects manufacturers to demonstrate system-level cybersecurity risk management across the full product lifecycle.
Threat modeling and Security Design Reviews (SDRs) are how these expectations are met. When performed early, SDRs identify architectural risks before they become embedded in regulated products, reducing rework and strengthening submission readiness.
In practice, however, manual, expert-dependent SDRs do not scale.
This whitepaper presents a scalable operating model for SDRs using automation, while preserving regulatory control. Automated SDRs perform consistent, first-pass analysis of design and architecture artifacts, surfacing system-level threats, assumptions, and risk scenarios that directly feed structured threat models.
Human experts retain authority over threat validation, risk acceptance, and mitigation decisions. SDR outputs are aligned to internal policies and quality systems, producing audit-ready, Design History File (DHF)– traceable evidence.
The result is scalable, repeatable threat modeling informed by design-time reviews, meeting FDA expectations without slowing development or increasing recall risk.
Why Security Reviews Matter in Medical Devices
Modern medical devices depend on software to perform safety-critical functions. Whether embedded firmware, cloud-hosted analytics, mobile companion applications, or Software as a Medical Device (SaMD), software behavior now directly influences clinical outcomes and patient safety.
This materially changes the effective attack surfaces beyond the physical devices, including:
Network interfaces and protocols
Cloud services and APIs
Mobile applications and authentication flows
Update and patch delivery mechanisms
Third-party software and infrastructure dependencies
This implies cybersecurity failures are no longer limited to confidentiality breaches. They can lead to:
Compromise of device availability or integrity
Unsafe device behavior under adversarial conditions
Product recalls or field safety corrective actions
Regulatory enforcement or delayed approvals
Loss of market authorization in one or more jurisdictions
Direct patient safety impact due to compromised device availability, integrity, or behavior
As a result, the FDA now expects manufacturers to demonstrate that cybersecurity risks are identified, assessed, mitigated, and monitored across the full product lifecycle, not addressed reactively.
Relevant requirements include:
FDA premarket and postmarket cybersecurity guidance
FDA Premarket Submissions for Management of Cybersecurity in
Medical Devices
Quality System Regulation (QSR) and ISO 13485 design controls
IEC 62304 software lifecycle requirements
IEC 81001-5-1 cybersecurity risk management for health software
HIPAA obligations where protected health information is processed
European Union Medical Device Regulation (EU MDR) expectations for globally marketed devices
Across these frameworks, the expectation is consistent: manufacturers must show system-level cybersecurity risk analysis, grounded in realistic threat assumptions, and integrated into design controls.
SDRs as Scalable Threat Modeling
Threat modeling is the primary mechanism by which these expectations are met in practice.
However, in regulated environments, threat modeling cannot be an informal exercise. It must:
Be repeatable across products and releases
Be performed early enough to influence architecture
Produce evidence suitable for audits and submissions
Cover the full system, not just individual components
SDRs operationalize threat modeling at the architecture level. A well- executed SDR evaluates how design choices introduce or mitigate risks related to:
Data flows and trust boundaries Authentication and authorization models Update, rollback, and recovery mechanisms Dependency and integration points Assumptions about the operating environment
When applied early, SDRs function as a preventive safety control, reducing the likelihood that insecure architectural patterns become embedded in regulated products.
Automated SDRs extend this model by enabling consistent threat identification across all designs, while retaining human oversight for:
Risk acceptance decisions
Selection of mitigations
Regulatory interpretation and justification
Crucially, automation does not replace expert judgment: human reviewers retain authority over risk acceptance, mitigation decisions, and regulatory interpretation. This directly supports FDA expectations for systematic, documented threat modeling while addressing the scale problem inherent in manual approaches.
Why Security Reviews Don’t Scale Today
SDRs are widely recognized as necessary in medical device development, yet they consistently fail to scale. The issue is not intent or awareness, but a structural mismatch between regulatory expectations for threat modeling and how design reviews are performed in practice.
Regulatory guidance emphasizes threat modeling as the mechanism for identifying system-level risks, assumptions, and mitigations. In real-world medical device development, this threat modeling is carried out through SDRs, where architecture and design decisions are evaluated for cybersecurity risk.
Regulatory guidance emphasizes threat modeling as the mechanism for identifying system-level risks, assumptions, and mitigations. In real-world medical device development, this threat modeling is carried out through SDRs, where architecture and design decisions are evaluated for cybersecurity risk.
Today, most SDRs, and therefore most threat modeling, remain manual and expert-dependent. Review quality varies with individual experience of FDA and IEC standards. Similar designs receive different findings, capacity is constrained by a small number of specialists, and consistency degrades as device portfolios grow.
At the same time, threat modeling expectations have expanded. Reviews are expected to identify system-level risks and mitigations, document pre- and post-mitigation risk states, state environmental assumptions (such as hostile hospital networks), and cover the full device lifecycle from supply chain and deployment to updates, interoperability, and decommissioning. Performing this level of threat modeling manually through SDRs for every design iteration does not scale.
These limits surface clearly in audits. Reviewers rarely find that SDRs were missing; instead, they see inconsistencies and weak traceability in the threat modeling outputs produced through those reviews. Threat models derived from SDRs differ across similar architectures.
Environmental assumptions are implicit or undocumented. Design risks identified during SDRs are not consistently reflected in cybersecurity risk management files. Late-stage risk acceptances reference schedules rather than technical justification, and lifecycle risks, particularly around updates and interoperability, are underdeveloped.
Timing compounds the problem. SDRs often occur after architecture decisions are already locked, turning findings into rework or forced risk acceptance. In this context, the threat modeling artifacts generated from SDRs become retrospective justification rather than design inputs.
Outsourcing SDRs and threat modeling provides temporary expertise but introduces cost, delay, poor fit for iterative change, and no sustainable internal review capability.
The impact is felt across teams. Development teams experience late feedback and documentation churn. Security and quality teams are forced to prioritize, leaving gaps in coverage and relying on tribal knowledge. Everyone agrees SDRs are necessary.
The problem is that manual, expert-driven SDRs cannot scale to produce consistent, regulator-ready threat models for modern medical device development.
The Seezo Approach: Automation That Preserves Your Process
Medical device organizations do not struggle with the concept of threat modeling, they struggle with applying it consistently, early, and at scale without breaking development processes.
Seezo is designed to address this gap. It scales threat modeling and SDRs consistently without changing how devices are designed, documented, or approved.
Seezo does not replace human judgment. In regulated environments, decisions about risk acceptance, mitigation sufficiency, and residual risk are inherently contextual and must remain under expert control.
Instead, Seezo automates the analytical groundwork that consumes most review time:
Extracting architectural context from fragmented documentation
Identifying relevant threat scenarios based on system design
Applying consistent security and threat-modeling logic across designs
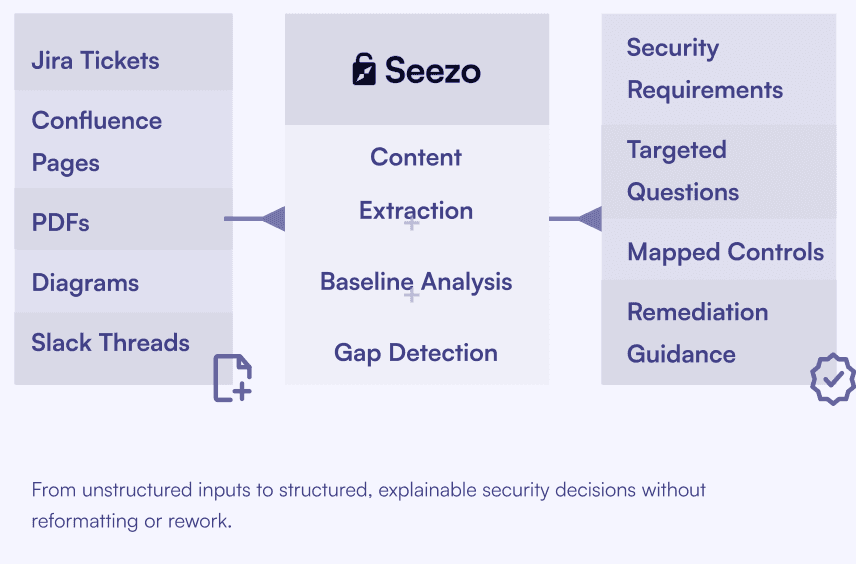
Medical device design information rarely arrives as clean diagrams or standardized templates. Architecture details are spread across
Product Requirements Documents (PRDs), system specifications, PDFs, diagrams, Jira tickets, Confluence pages, and regulatory documentation. Seezo ingests these existing artifacts directly and analyzes them as-is. It extracts security-relevant contexts such as data flows, trust boundaries, interfaces, update mechanisms, and external dependencies without requiring teams to re-document designs for security purposes.
Seezo applies a large set of predefined security rules aligned to medical device–specific risk patterns, including areas emphasized in regulatory guidance:
Authentication and authorization models for devices, services, and users
Data integrity and availability risks affecting device behavior
Update, rollback, and recovery mechanisms
Trust boundaries between device, cloud, mobile, and third-party
systems
Assumptions about deployment environments (e.g., hostile hospital
networks)
Supply-chain and integration-related risks
This ensures threat modeling is systematic and repeatable, rather than dependent on individual reviewer intuition.
With Seezo in place:
Security and quality teams can review more designs without increasing headcount Reviews can occur earlier, before architecture decisions are locked Development teams receive clearer, more actionable feedback tied to internal standards Coverage expands from selective (only “high-risk” designs) to systematic, repeatable review of all design changes Aligned with medical device development workflow supporting patient safety and regulatory expectations without slowing delivery or adding process overhead SDR findings feed directly into system-level threat models, ensuring consistent identification of threats, assumptions, and mitigations across products and releases
Customization: Your Rules, Your Standards
Medical device regulatory guidance defines expectations, but compliance depends on how those expectations are interpreted, implemented, and justified within an organization. SDRs must therefore align to internal controls, not abstract best practices.
Seezo operates within an organization’s existing security architecture, quality system, and regulatory interpretations, ensuring SDR outputs are usable as formal design-control evidence.
Customization starts by configuring Seezo against:
Internal security standards (e.g., identity models, encryption requirements, update and rollback controls)
Quality system procedures governing design inputs, risk analysis, and design reviews
Established regulatory interpretations previously accepted in submissions and audits
During onboarding, Seezo is calibrated to organization-specific terminology, system boundaries, and architectural patterns. For example, how the organization defines a “system,” “component,” or “interface,” and how trust boundaries are drawn between device, cloud, mobile, and third- party services.
Security findings are generated by applying predefined rules that encode both medical device–specific risk patterns and org-specific control requirements
Each finding is explicitly linked to:
The architectural element or data flow that triggered it
The internal policy or control requirement being evaluated
The design decision or assumption under review
This allows reviewers to determine whether:
A control is met
A mitigation is required
Residual risk is acceptable under internal risk criteria
Risk acceptance and exceptions are captured with rationale, supporting defensible decisions rather than informal approvals. For external alignment, findings can be mapped to:
U.S. FDA cybersecurity guidance
IEC 62304 software lifecycle requirements
IEC 81001-5-1 cybersecurity risk management expectations
ISO 13485 design control clauses
STRIDE threat modeling categories
OWASP Top 10, where applicable
These mappings provide evidence linkage, not prescriptive direction. Internal controls remain the source of truth.
This level of customization ensures:
SDR outputs can be referenced directly in regulatory submissions
Findings align consistently across architecture documents, risk files,
and design reviews
DHF traceability from architecture → threat → mitigation → acceptance
Audit responses rely on recorded rationale, not reviewer memory
Workflow Automation — End-to-End Integration

SDRs struggle to scale in medical device organizations due to workflow friction, not lack of intent. Fragmented design systems, manual reviews, and disconnected feedback cause delays, lost context, and repeated rework as designs evolve.
SDRs struggle to scale in medical device organizations due to workflow friction, not lack of intent. Fragmented design systems, manual reviews, and disconnected feedback cause delays, lost context, and repeated rework as designs evolve.
Seezo addresses this by embedding SDRs directly into development workflows. Reviews are initiated from existing design artifacts such as architecture documents, specifications, diagrams, and tickets without requiring security-specific templates or parallel documentation. As designs change, updated artifacts can be re-reviewed incrementally, preserving continuity and traceability.
Security feedback is delivered back into the same systems teams use to manage design decisions and remediation work. Developers see security findings alongside other design considerations, reducing context switching and making feedback actionable at the point of decision.
Seezo integrates with tools commonly used in medical device development, including Confluence, Jira, Google Drive, SharePoint, Notion, Slack, and ServiceNow, with planned support for GitHub, Miro, and Lucidchart. Custom integrations are available to align SDR outputs with regulated quality systems and documentation repositories.
Importantly, automation does not bypass quality controls. Review checkpoints, approvals, and sign-offs remain aligned with established quality system requirements, while inputs, findings, and decisions are captured automatically. The result is an SDR process that scales with development velocity, generates audit-ready evidence and avoids workflow disruption or process risk.
Key Benefits Summary
For Product Security, Quality, and Regulatory Leadership
Seezo makes SDRs a consistent and reliable design control rather than an expert-driven activity. Each review result is traceable to the device architecture, applied threat-model logic, and the internal security and design controls used to evaluate it.
Seezo helps teams determine whether a device design meets internal control expectations, residual risks, and what mitigations are required before design freeze. Risk acceptance and exceptions are documented with evidence, aligning with FDA cybersecurity expectations and internal risk management practices.
Since Seezo integrates into existing development workflows, leaders gain visibility into review coverage and outcomes across devices without adding new tools or workflows. This reduces dependence on individual expertise and improves consistency across products.
For Development Teams
Development teams receive security feedback within their existing design and documentation tools. Feedback is specific to the device architecture and intended use, tied directly to internal standards and assumptions.
Teams understand what design choices introduce risk and what changes are required to meet organizational expectations. Automated baseline analysis provides feedback early while architecture and interfaces are still flexible. This reduces late-stage design changes, limits disruption to verification and validation activities, and avoids security-driven rework close to submission milestones.
For the Organization
Seezo SDRs scale with product complexity and release velocity. Reviews are applied consistently across devices, software updates, and product lines, including connected devices and SaMD.
Outputs align to internal design controls and map to FDA cybersecurity guidance, ISO 13485, IEC 62304, IEC 81001-5-1, and STRIDE. This supports audit readiness and submission consistency without additional manual effort.
Early identification of architectural risks reduces postmarket security findings, recall likelihood, and regulatory disruption, while preserving development velocity and patient safety.
Evaluate Seezo in your medical device development workflow
See how design-time security decisions can be applied consistently across a high volume of architectural changes without adding headcount or changing developer workflows.
Get Started with Seezo
Have questions or want to learn more about how Seezo can help your team? Reach out to us directly: hi@seezo.io
Experience the benefits of Seezo SDR firsthand. Book a walkthrough.